Cystic Fibrosis - Symptoms, Causes, Risk factors, Complications, Treatment & Prevention
PACE Hospitals
Cystic fibrosis definition
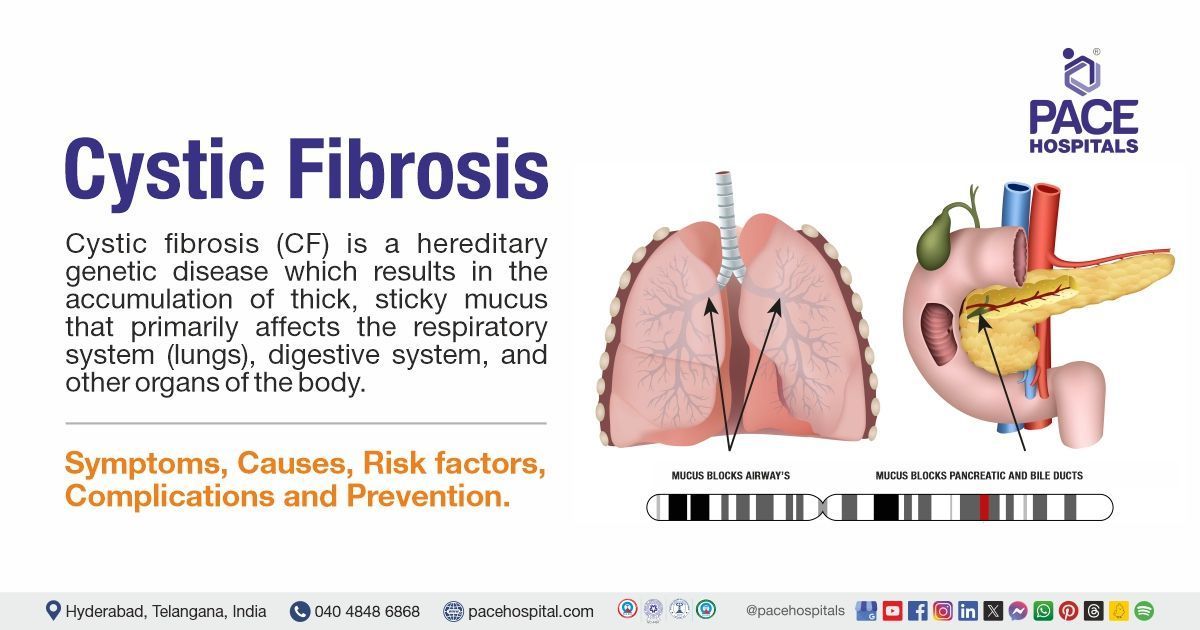
Cystic fibrosis (CF) is a hereditary genetic disease that results in the accumulation of thick, sticky mucus which primarily affects the respiratory system (lungs), digestive system, and other organs of the body.
When individuals have CF, their mucus becomes extremely thick and sticky. People with cystic fibrosis have difficulty clearing this mucus from their lungs. It blocks the small airways and traps bacteria, leading to recurrent infections and obstructions, which can result in permanent lung damage as time passes.
Thick mucus in the digestive tract can impair the digestive enzymes flow from the pancreas to the small intestine, making it difficult to digest fats and absorb certain nutrients.
Cystic fibrosis disease is typically managed by a team of healthcare experts, primarily including a
pulmonologist (lung specialist), because it primarily affects the lungs. Other specialists include a
gastroenterologist who treats CF-related digestive issues, such as malabsorption and
pancreatitis.
Cystic Fibrosis Prevalence
Prevalence of cystic fibrosis worldwide
A 2022 study found that an estimated 1,62,428 people globally have cystic fibrosis (CF), with 65% being identified and 12% receiving triple combination medications. Additionally, 57,076 people remain undiagnosed.
Prevalence of cystic fibrosis in India
In India, the prevalence of cystic fibrosis isn't well established, Only 600 people have been officially diagnosed with cystic fibrosis (CF), however this figure is far lower than the actual prevalence due to underdiagnosis and a lack of awareness. Approximately 19% to 34% of Indian CF patients carry the DF508 mutation, which is one of the most frequent genetic variants associated with CF.
Cystic Fibrosis Classification
Cystic fibrosis is often classified based on the mutations, severity, and presentation of the disease, particularly in terms of symptoms and how early or severe the disease manifests, including:
CFTR mutations can be classified as per mechanisms that disrupt CFTR function
- Class I mutations: Defective protein production
- Class II mutations: Defective protein processing
- Class III mutations: Defective regulation
- Class IV mutations: Defective chloride conduction
- Class V mutations: Reduced amounts of functional CFTR protein (less transcription)
These classes are classified based on the severity of the condition, such as:
- Class I, II and III usually lead to a classic (severe) CF phenotype with pancreatic insufficiency.
- Class IV and V are mostly associated with a milder expression of the disease.

Cystic Fibrosis Symptoms
Signs and symptoms of cystic fibrosis typically appear in early childhood; however, they can also occur shortly after birth or later in adulthood. Some of the main symptoms of cystic fibrosis, according to the affected organ, are listed below:
Cystic fibrosis in lungs symptoms
There are many signs and symptoms of cystic fibrosis. Some are related to the respiratory system when the mucus clogs block airways. They are
- Ongoing chronic coughing
- Shortness of breath
- Wheezing
- Frequent sinus infections
- Frequent lung infections
- Clubbing or enlargement of the fingertips and toes
Cystic fibrosis digestive system symptoms
Cystic fibrosis affects the digestive system. Many people experience digestive difficulties caused by the pancreas failing to function properly. Some common cystic fibrosis digestive symptoms include:
- Difficulty gaining weight
- Poor growth
- Bowel obstructions
- Frequent greasy or oily bowel movements
Other cystic fibrosis symptoms in adults may include:
- Fertility problems
- CF-related diabetes
- Urinary Incontinence
- Low bone mineral density
Cystic fibrosis symptoms in kids include lung infections, salty-tasting skin, daily cough, at times with mucus, poor growth or slow weight gain even with a good appetite and shortness of breath, and trouble having a bowel movement.

Cystic Fibrosis Causes
Cystic fibrosis is only caused by a mutation in the gene called cystic fibrosis transmembrane conductance regulator (CFTR), which regulates the production of the CFTR protein. The following are the types of cystic fibrosis mutations that cause it:
Types of mutation in cystic fibrosis
There are about 2000 mutations in the CFTR gene that can cause cystic fibrosis. These mutations are grouped into five categories:
- Defective protein synthesis: This mutation fails to transform the genetic information into a protein product, resulting in a complete absence of the CFTR protein.
- Defective protein processing: This malfunction causes the CFTR protein to undergo abnormal post-translational processing. As a result, CFTR cannot be moved to the proper cellular site.
- Disordered regulation: This malfunction leads to decreased protein activity in response to intracellular signals. As a result, protein channels are completely non-functionally formed in the cell.
- Defective chloride conductance: This malfunction occurs when the protein is generated and properly located on the cell surface, but CFTR Protein works slowly because the rate of chloride ion flow decreased than normal.
- Accelerated channel turnover: This malfunction occurs when there is insufficient CFTR protein in the cellular membrane due to fast breakdown by cellular processes.
The result of all mutations indicates that the production of CFTR protein does not function properly and disturbs the movement of salt and water in the cells. These changes lead to the buildup of thick mucus in the lungs, digestive system, and other organs in the body.
Cystic fibrosis inheritance pattern
Everyone inherits two CFTR genes, one from the mother and the other from the father. Children who inherit a CFTR gene with a mutation from both parents will develop cystic fibrosis.
A cystic fibrosis carrier is a person who inherits a mutant CFTR gene from one parent and a normal CFTR gene from the other parent. Cystic fibrosis carriers can pass the mutant CFTR gene to their children. Cystic fibrosis gene carriers are normally healthy but may experience mild symptoms.

Cystic Fibrosis Risk Factors
Identifying and addressing these cystic fibrosis risk factors, helps in the diagnosis and management. The following are the cystic fibrosis risk factors:
- Family history: One of the leading risk factors for cystic fibrosis (CF) is having a family history of the condition, which means it is inherited when both parents carry the faulty CF genes as it can be passed to the next generation.
- Cystic fibrosis genetics: Cystic fibrosis is an autosomal recessive disease, meaning individuals must inherit two defective copies of the CFTR gene, one from each parent, to develop the condition.
- Ethnicity: CF can affect anyone, and the specific gene mutations that cause CF also differ according to geographic region and ethnic background. Some ethnicities have a higher carrier rate of the cystic fibrosis gene mutation, which increases the risk of passing on the condition.

Cystic Fibrosis Complications
Long-term complications of cystic fibrosis can arise when the condition is left untreated, which are listed below:
- Diabetes: Cystic fibrosis causes diabetes because the mucus buildup results in inflammation and scarring of the pancreas, which prevents it from producing sufficient insulin, resulting in increased blood glucose levels.
- Cardiovascular (heart) disease: Cardiac dysfunction in CF is not only a complication of pulmonary hypertension. It is caused by the destruction of lung parenchyma and additional factors, including CFTR gene polymorphism, systemic inflammation, hypoxia-induced myocardial damage, diabetic cardiomyopathy, and a lack of certain essential trophic factors, such as proteins or molecules that promote the survival, growth, and differentiation of cells.
- Colorectal cancer: Colorectal cancer is a complication of CF, but the exact cause is not fully understood. Cystic fibrosis affects the digestive system, leading to thick mucus, causing intestinal blockages, rectal prolapse and inflammation. This inflammation may increase the risk of colorectal cancer.
- Pneumonia: Pneumonia is a complication of cystic fibrosis because the mucus in the lungs is thicker, sticky, and more difficult to clear, leading to the trapping of microorganisms such as bacteria, viruses, or, in rare cases, fungi, resulting in a high likelihood of repeated chest infections.
- Infertility: Men with cystic fibrosis have a congenital bilateral absence of the vas deferens (CBAVD), a condition called azoospermia in cystic fibrosis (male infertility in cystic fibrosis) where the sperm tubes are absent, which prevents sperm from passing from the testicles to the urethra.
- Osteoporosis and arthritis: Some of the factors that contribute to bone disease in the cystic fibrosis population include decreased absorption of fat-soluble vitamins (particularly D and K) due to pancreatic insufficiency, delayed puberty because of altered sex hormone production, chronic inflammation, a lack of physical activity, glucocorticoid treatment, and intrinsic hyper-resorptive bone physiology (imbalance in the bone remodeling process). In other studies, 75 percent of adult individuals with CF had osteopenia (loss of bone density) or osteoporosis.
- Liver diseases: Cystic fibrosis can cause liver impairment by accumulating mucus and blocking bile ducts. This prevents bile from leaving the liver and results in inflammation and scarring (fibrosis), which in turn results in abnormal liver function.
- Pneumothorax: The main cause of pneumothorax in CF is the damage and structural changes in the lungs, which make it easier for air to leak into the chest.
- Malnutrition: Cystic fibrosis causes malnutrition because the condition causes thick mucus to obstruct the pancreas and prevents digestive enzymes from reaching the intestine, known as malabsorption, which makes it difficult for CF patients to absorb nutrients and digest food.
Cystic Fibrosis Diagnosis
Cystic fibrosis diagnosis usually involves a single test or a combination of tests performed by a healthcare professional to confirm the presence of the disease:
- Medical history
- Physical examination
- Newborn screening
- Newborn blood spot test
- Confirming cystic fibrosis
- Sweat test
- Genetic test
- Screening and preconceptional diagnosis of cystic fibrosis
- Cystic fibrosis carrier testing
- Prenatal cystic fibrosis screening
- Chorionic villus sampling (CVS)
- Amniocentesis
- Chest x-ray
- Sputum culture
- Stool test
Cystic Fibrosis Treatment
Cystic fibrosis treatments help in managing symptoms and slow down lung damage as it has no cure. As a result of various treatment options, many cystic fibrosis people are living healthier lives. Below are some of the cystic fibrosis treatment options:
Conservative management (non-surgical treatments)
- Airway clearance techniques
- PEP therapy
- High‐pressure PEP therapy
- Active cycle of breathing techniques
- Autogenic drainage
- Airway oscillating devices
- Cystic fibrosis medications
- Antibiotics
- Anti-inflammatory medicines
- Bronchodilators
- CFTR modulators
- Mucus thinners
- Breathing support
- Oxygen therapy
- Pulmonary rehabilitation
- Gene therapy
- Dietary management
- Exercise
Cystic fibrosis Surgical management
- Ventilator support
- Extracorporeal membrane oxygenation (ECMO)
- Lung transplant
Why Choose PACE Hospitals?
Expert Super Specialist Doctors
Advanced Diagnostics & Treatment
Affordable & Transparent Care
24x7 Emergency & ICU Support
Cystic Fibrosis Prevention
Cystic fibrosis is a genetic condition; hence, it cannot be prevented. However, one can reduce the risk of passing it to the child by taking the following genetic tests to detect mutated CFTR genes before and during pregnancy. If someone is planning on becoming a parent, as mentioned. The following are some of the genetic screening and counselling techniques that can help to manage the risk:
Cystic fibrosis Genetic Counseling
- Preconceptional test: It is done before pregnancy to determine whether an individual (or couple) is a carrier of the CFTR gene mutations that cause cystic fibrosis
- Prenatal test: It is performed during pregnancy and can identify whether a fetus is at risk for CF.
- Genetic counselling for cystic fibrosis patients: Genetic counsellors are medical professionals qualified in both genetics and counselling as well as they provide emotional support to CF patients and help them to understand what it is, what causes it, its inheritance and what diagnostic tests can help to detect mutated genes, finding the risk of passing this condition to the child.
Cystic fibrosis patient education
Preventing the spread of transmissible infections in CF patients: It is a top priority in caring for individuals with cystic fibrosis (CF). The following are some preventive measures that help avoid infections in cystic fibrosis patients:
- Following hand hygiene
- Maintaining segregation
- Using facemasks
- Enhanced adherence (encouraging and helping patients stick to their prescribed medications, therapies, or lifestyle changes).
Difference between Pulmonary Fibrosis vs Cystic Fibrosis
Pulmonary fibrosis vs cystic fibrosis
Pulmonary fibrosis and cystic fibrosis are both disorders that can impair the lungs and make breathing difficult, but they differ in several ways, as detailed below:
| Elements | Pulmonary fibrosis | Cystic fibrosis |
|---|---|---|
| Causes | Pulmonary fibrosis is caused by a buildup of scar tissue in the lungs | Cystic fibrosis occurs when a mutation in the CFTR gene is inherited autosomally recessive. |
| Symptoms | Symptoms of pulmonary fibrosis are dry cough, shortness of breath and weight loss. | Symptoms of pulmonary fibrosis are dry cough, shortness of breath and weight loss. |
| Life expectancy | The typical survival time for pulmonary fibrosis is three to five years, but some people live far longer. | Cystic fibrosis patients who reach adulthood have an average life expectancy of over 50 years. |
Difference between COPD and Cystic Fibrosis
COPD vs cystic fibrosis
Cystic fibrosis (CF) and
chronic obstructive pulmonary disease (COPD) are both lung disorders that cause airway blockage and inflammation, although they differ in various ways, including:
| Elements | COPD | Cystic fibrosis |
|---|---|---|
| Causes | COPD is caused by environmental causes, including cigarette smoking and on genetic basis. | Cystic fibrosis is a genetic disorder occurred by mutations in the CFTR gene. |
| Symptoms | COPD symptoms include trouble breathing, wheezing, chronic cough, chest tightness, and frequent lung infections. | CF symptoms include salty skin, diarrhoea, and chronic lung symptoms such as wheezing, coughing, and thick mucus. |
| Age of diagnosis | COPD is usually diagnosed after age 40. | CF is usually diagnosed early in life |
Frequently Asked Questions (FAQs) on Cystic fibrosis
Is cystic fibrosis curable?
Cystic fibrosis (CF) has no cure, but treatment can help people live longer and healthier lives. Treatments include medications, airway clearance techniques, nutritional support, breathing support, and surgery, such as lung or pancreas transplants if organs are severely damaged. These treatments help treat chest infections, thin mucus, widen airways, loosen mucus, and provide a supportive environment for overall health.
Is cystic fibrosis a genetic disorder?
Cystic fibrosis (CF) is a genetic disorder caused by a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. This mutation alters the CFTR protein, controlling cell movement. CF patients inherit two mutated copies of the CFTR gene (one) from each parent, requiring both parents to have at least one defective gene.
What is the life expectancy of someone with cystic fibrosis?
The cystic fibrosis life expectancy has significantly improved in recent years, with a median survival age predicted to be almost 50 years. Factors leading to this improvement include advancements in treatments, new genetic advancements for diagnosis and treatment, and earlier diagnosis.
Are there prenatal tests for cystic fibrosis?
Yes, chorionic villus sampling (CVS) and amniocentesis are prenatal diagnostic tests for cystic fibrosis and other diseases. CVS is done between 10 to 13 weeks of pregnancy, and amniocentesis is often performed between 15 and 20 weeks of pregnancy. However, it can be done later until birth.
What is cystic fibrosis 6 foot rule?
The 6-foot rule refers to maintaining a physical distance of at least 6 feet from others because it is the distance germs can travel when a person coughs or sneezes. Some germs and bacteria are considered particularly dangerous for individuals with CF, as they can to spread to one another.
What is cystic fibrosis?
Cystic fibrosis is a genetic condition affecting a protein in the body, causing thick and sticky mucus. This condition affects the body's cells, tissues, and mucus-making glands. Normal mucus protects the airways, digestive tract, and other organs, but cystic fibrosis causes it to become thick and sticky, potentially causing blockages, damage, or infections.
Is cystic fibrosis dominant or recessive?
Cystic fibrosis (CF) is a recessive disease characterized by mutations in both CFTR gene copies. CF is inherited in an autosomal recessive manner, which means both copies of the gene in each cell carry mutations. Therefore, a carrier does not develop the condition.
How does cystic fibrosis affect sweat glands?
Cystic fibrosis (CF) affects sweat glands by causing sweat that is high in salts, increased sweating, and reversed chloride flow. People with CF have high chloride levels, which can make their skin feel salty. They may sweat more than those without CF, and the chloride channel doesn't reabsorb chloride, leading to sodium loss and fluid loss. The amount of chloride in sweat is often used to diagnose CF, which involves stimulating a small area of skin to secrete sweat.
Which chromosome causes cystic fibrosis?
A gene called the CFTR (cystic fibrosis transmembrane conductance regulator) that is located on the seventh pair of chromosomes causes cystic fibrosis.
What is the role of salt in cystic fibrosis?
Salt, often known as sodium chloride, helps muscles contract. Inadequate salt intake can inhibit growth, reduce appetite, and cause stomach pain, weakness, muscle cramps, nausea, and headaches. Individuals with CF lose a lot of salt through sweat. Therefore, they must eat salty meals, especially in hot, humid conditions.
What kind of diet for cystic fibrosis?
People with CF have estimated energy needs that are 1½ to 2 times higher than those without the condition. A high-calorie, high-fat diet is often recommended, with fat accounting for 40% of total calories.
What causes thick mucus in cystic fibrosis?
A faulty gene known as the cystic fibrosis transmembrane conductance regulator (CFTR) causes thick mucus to accumulate in cystic fibrosis. This CFTR gene regulates water and salt movement into and out of the body's cells. These changes produce thick and sticky mucus.
What role do goblet cells play in cystic fibrosis?
Goblet cells contribute to cystic fibrosis (CF) by producing mucins, the primary protein components of mucus. In CF, goblet cells show abnormal mucus production and retention, which can lead to an accumulation of mucus in the airways.
Is cystic fibrosis polygenic?
Cystic fibrosis is commonly thought to be a single-gene disease caused by recessive mutations in the CFTR gene, which is often classified as "oligogenic" rather than polygenic because it involves few genes.
When to see a doctor for cystic fibrosis?
The lungs and digestive system are affected by the hereditary disorder cystic fibrosis. Some people, particularly those with milder forms, may initially have mild symptoms mistaken for common digestive or respiratory issues. Ignoring signs can cause long-term organ damage, recurrent infections, and a delay in diagnosis.
Key warning signs:
- Persistent cough with thick mucus
- Frequent chest or lung infections
- Shortness of breath or wheezing
- Poor weight gain
- Bulky, greasy, or foul-smelling stools
- Abdominal bloating or constipation
- Salty-tasting skin
- Fatigue or reduced stamina
If these symptoms start early in childhood or persist over time, see a pulmonologist or cystic fibrosis doctor who treats cystic fibrosis. Early identification lowers complications, enhances nutrition and lung function, helps initiate therapy on time, and dramatically improves long-term health results.
Share on
Request an appointment
Fill in the appointment form or call us instantly to book a confirmed appointment with our super specialist at 04048486868
Appointment request - health articles
Recent Articles